A Focus on Computational Tools & Data at the ACS Spring National Meeting

Spring is in the air! It was exciting for chemists to assemble in person at the Spring National American Chemical Society (ACS) Meeting in Indianapolis, March 26-30. This year, it was a hybrid event supporting both in person and virtual participation. In person attendance was high, with more than 10,000 attendees. There were an additional 1,697 virtual attendees representing 68 countries.
One of the themes of the ACS meeting was the expanding role computational methods and tools play in chemistry discovery, both in the discovery of new pharmaceuticals and new materials. Also explored in the meeting was the growth of analytics, chemoinformatic and statistical tools to deal with the ever-growing abundance of chemical data information.
Here is a quick look at some of the sessions, presentations and symposia at ACS that explored this growing role of computational tools in chemistry, and their use in drug design.
Revvity Signals sponsored a Division of Chemical Information (CINF) symposium. Rachelle Bienstock chaired the symposium and presented a talk entitled “Electronic Lab Notebooks and Notebook-Like Applications for Data Sharing.” Her session explored new electronic notebooks and cloud based collaborative data sharing tools.
Cheminformatics, Electronic Notebooks, and Laboratory Information Management Systems
This session was devoted to chemistry database Application Programming Interfaces (APIs), available databases, workflows and use cases, and the use of REST or RESTFUL APIs to formulate queryable use URLs in web browsers.
Presentations were given on programmatic access to PubChem, ChEMBL, UniChem, and CAS (Chem Abstracts Services). IUPAC held a workshop on Advancing Fair Chemistry: Developing New Services focused on sharing chemical data. The workshop also included a discussion on developing standardized programmatic access to chemical information (protocol services).

Another symposium was held on cheminformatics and computational-based chemical library enumeration. Presentations were given on efficient methods to explore large chemical space in enumerating chemical compound libraries for virtual screens, including chemical structure validation and global chemical resolver. One intriguing presentation from International Flavors and Fragrances focused on the design of chemical libraries for fragrance design – a method similar to those used by medicinal chemists.
Chemists packed a symposium on electronic lab notebooks and notebook-like applications for data sharing. This featured presentations on collaborative data sharing tools. There were also presentations by academic groups (NFDI4Chem, a consortium formed within the German national research data infrastructure, and the UK Physical Sciences Data infrastructure), and Revvity Signals spoke on the new Signals OneTM, focusing on the Make-Test-Decide workflow for drug discovery.
New Drug Design Modalities
The medicinal chemistry division of the ACS is one of the largest divisions, and the national meeting is an opportunity for the presentations of “first disclosures” – including those compounds most recently approved as well as presentations of new compounds and drug design ideas.
First Disclosures of New Drugs Entering the Clinic
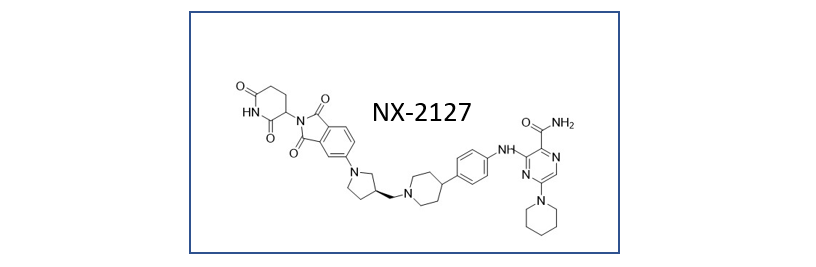
Nurix Therapeutics clinical candidate NX-2127 targets Bruton’s Tyrosine Kinase (BTK) and degrades BTK for the treatment of B-cell malignancies. This compound is a combined BTK degrader and an Ikaros zinc finger transcription factor degrader combined by a linker.
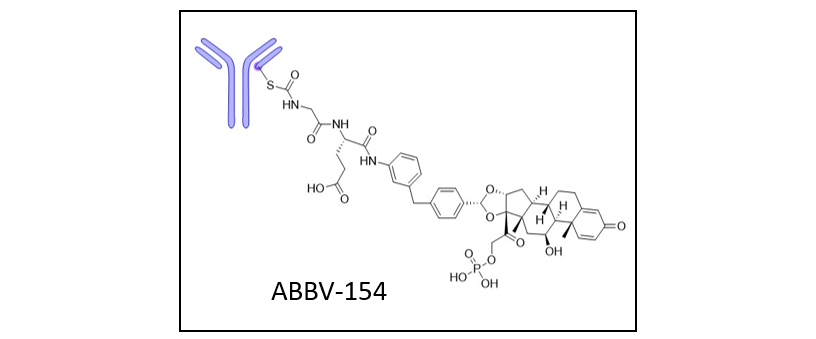
AbbVie presented ABBV-154, a compound to treat Crohn’s disease and polymyalgia rheumatica by inhibiting the tumor necrosis factor. ABBV-154 connects an antibody to an antibody-drug conjugate (ADC) using a glucocorticoid receptor modulator (GRM) as a linker.
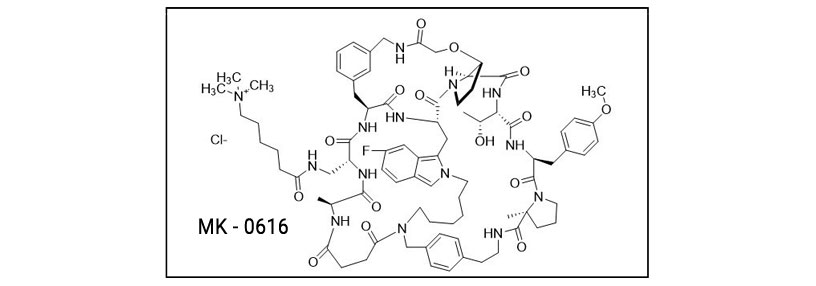
Merck presented MK-0616 targeting PCSK9 for the treatment of heart disease. This drug is a macrocyclic peptide which was designed using structure-based drug design methods.
Boehringer Ingelheim presented 1015550 a Phosphodiesterase 4 inhibitor for the treatment of Idiopathic pulmonary fibrosis.
Machine Learning in Drug Design
Another main theme at the ACS meeting was the increasing incorporation of Machine Learning (ML) techniques in computational drug design methods. It was exciting to see many symposia honoring the accomplishments of individuals in computational and structure-based drug design.
One presentation discussed the use of Alpha Fold2 – an artificial intelligence deep learning program developed by DeepMind – to predict target protein structures for a GPCR (G-coupled protein receptor). The protein target model developed was then used, in conjunction with induced fit docking and molecular dynamics, successfully for ligand design. Five Alphafold2 models of the GPCR were used in combination to sample the helix rotation within the transmembrane helix bundle to obtain a useful model for the docking studies.
Professor Charlotte Deane MBE, Oxford University posed the question, “Can machine learning understand the physics of binding?” and described the PointVS ML software designed by her group which identifies plausible ligand-binding interactions.
Dr. Koes, University of Pittsburgh presented GNINA (pronounced “NINA”), a convolutional neural network 3D grid which can perform whole protein and flexible ligand docking. This software performed well on cross-docked protein ligand docking scoring and demonstrated use in the virtual screening of ligands.
Designing drugs which can pass through the blood brain barrier remains one of the biggest drug discovery challenges. Dr. Fabio Urbina presented a method for confident predictions of blood brain barrier crossing using ML models with conformal predictions and applicability constraints.
Computational and Structure-Based Drug Design Methods
Professor Bill Jorgensen (Yale) presented the use of free energy perturbation (FEP) calculation methods to shorten and enhance drug discovery times for HIV-RT drug design and SARS-Cov2 non-covalent Mpro inhibitors. Once thought to be too complex, too long, and inefficient for drug discovery projects, in specific drug discovery cases FEP is now able to predict binding affinities and is becoming a consistently applied technique prioritizing compound synthesis and enhancing the drug discovery process. He highlighted animal studies of some highly effective SARS-Cov2 inhibitors.
The Open Force Field initiative is developing a new force field named Rosemary, slated for Q3 2023. It will support proteins, and work towards a facile polarizable electrostatic model, partial charges, and atom-centered point polarizability. This will enhance the applicability of FEP and other molecular dynamics calculations and studies for drug discovery.
The Growing Role of Computational Tools and Data Management in Drug Discovery
As you can see, presentations at the Spring 2023 ACS meeting highlighted a common theme – the importance of computational and data management methods in drug discovery. These methods play a pivotal role with some of the new drug modalities presented in the first disclosures, including molecular degraders, antibody-drug conjugates, covalent drugs and peptidic drugs.
Revvity Signals One can play a significant role in expediting drug discovery. Contact us today to learn more.